With the recent appearance of novel SARS-CoV-2 lineages in the UK, South Africa and Brazil, which are suspected to display increased infectivity and/or partial escape from neutralizing antibodies, the need for continuous tracking of viral evolution through genome sequencing has received increased public attention.
While the UK’s COVID-19 Genomics Consortium (COG-UK) has been pioneering such monitoring since early on in the pandemic, other countries are now eager to establish their own networks for genomic surveillance.
In the framework of covid19.galaxyproject.org, an international team of Galaxy developers and researchers from different disciplines (including members of the Galaxy EU team and the Institute of Virology of the University Medical Center Freiburg) have collaborated since February last year to establish a set of workflows for SARS-CoV-2 genomics analysis - including the ones that are urgently needed now.
The European Galaxy server offers these as public workflows. Just search the list for the tag covid19.galaxyproject.org and you will find our recommended, best-practice workflows for SARS-CoV-2 variant analysis from a variety of sequencing data sources: be it whole-genome sequencing or ampliconic (ARTIC) data sequenced using short-read (Illumina, single-end or paired-end) or long-read (ONT) data, we have workflows for calling variants (even low-frequency intra-sample ones), generating standardized technology-independent reports, and for building reliable consensus sequences!
All you need to bring is your sequencing data, everything else you will need as input to the workflows (SARS-CoV-2 reference genome, ARTIC primer scheme, etc.) is already made available for you through a public COVID-19 Resources history.
Our workflows have been built and tested specifically with scalability in mind: on usegalaxy.eu we have verified, using large batches of COG-UK and other data, that they let you analyze even thousands of samples in parallel.
Combine, e.g., our ARTIC Illumina PE variant calling workflow with the workflow for consensus sequence building and you have an analysis pipeline for analyzing typical diagnostic batches of data, like most of the data generated by COG-UK, and for turning the results into a set of consensus sequences ready for submission to GISAID or for clade assignment with Nextstrain.
If, instead of reducing all the precise variant-calling information into a simplifying consensus sequence, you would like to explore the full picture of potential intra-sample variation, you could run our variation reporting workflow, which will create different graphical and tabular views on the variant data, which you can use as a starting point for further investigation, e.g. with Interactive Notebooks or using resources like https://observablehq.com/collection/@spond/intrahost-variant-exploration.
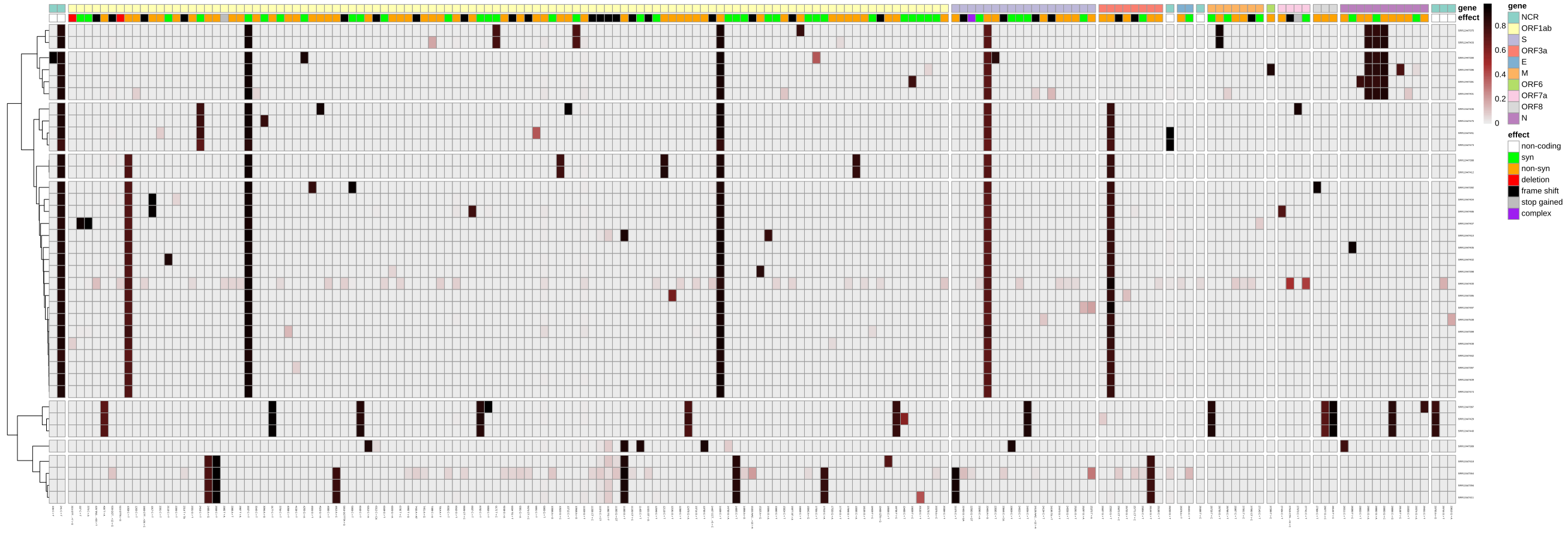
A workflow-generated plot showing the variants and their allele-frequencies identified across 38 Illumina-sequenced SARS-CoV-2 samples.
Finally, in order to ensure open access to original sequencing data, we stronlgy advocate, and offer tools and assistance, for submitting raw sequencing data to public archives like ENA - so that everyone can reanalyze the data and draw their own conclusions.
P.S.: If you want to know more about our COVID-19 activities, please have a look at our running document.